
How to BLAST your way to the truth about the origins of COVID-19
How to BLAST your way to the truth about the origins of COVID-19
Using BLAST is easy. I'm going to show you how easy and how to prove that SARS-Cov-2 is man-made

I’ve been meaning to write this blog for ever. Well, at least since Prashant Pradhan (a wonderful, honest and brave genomics scientist) raised the possibility back in February 2020 that the SARS-Cov2 virus was man made. And we have seen multiple confirmatory pieces that the virus was made in a lab, one of the better ones here on zenodo and with its own cute video for non-Bayesian peeps here. As of writing this those links are still up which at 12 months is pretty good going for any article that dares challenge the drivel propagandised by our beloved “free press [sponsored by pharma]”.
Anyway, BLAST is the NCBI/NIH (aka US government) repository for genomic and proteomic sequences, amongst other things. It is where all genome scientists around the world deposit their sequences if they make a discovery. Its main function is to allow comparison of gene sequences and discovery of sequences that match one that you might have come across in your experiment. What’s a gene sequence? That’s easy. It’s a line of code, made up of any combination of 4 letters in a sequence. Remember the film GATTACA? If you haven’t watched it by now, you should - because it’s yet another dystopian movie that is now too close to home.
 - AZ Movies")
The movie’s title is based on the 4 nucleotide bases (G, A, T, C) which make up the genetic code of every human’s DNA. There are about 3 billion of them in each cell, making a code that is unique - resulting in you, a unique individual! The code pairs up so that G-C and A-T always combine to make the double-helix that you see in the picture, such that GATTACA would be paired with CTAATGT (the complement). The code is read in a specific direction so GATTACA on one strand would be TGTAATC on the other (the reverse complement). One of the good things about BLAST is that it doesn’t care which version you give it, it will still point you to the correct gene.
One other thing to note at this point is probabilities. You did this at school with tossing a coin (where the code is H for heads or T for tails). What would be the probability of HHHH (1 in 2^4 = 1/16). The same applies for TTTT. The same applies for THTH, or any specific sequence of coin tosses. Try it yourself if you don’t believe me (predict the sequence first and then see how many times you have to run it). Genetic code is essentially a “four sided coin”. So for any run of a specific sequence (e.g. GATC) the probability of getting that EXACT sequence is 1 in 4^4, or for any number n of nucleotides (nt or bases) the chance is 1 in 4^n (this is simplified because in some situations the probability of the next base being X depends on the surrounding bases).
BLAST has two sections - nucleotide (BLASTn) and protein (BLASTp). BLASTp deals with amino acid sequences, in just the same way as nucleotide sequences. But there is a big difference because there are 20 amino acids (rather than 4 nucleotides) and therefore even short runs (e.g. QTNS = Glu-Thr-Asn-Ser) would carry a probability of somewhere around 1 in 20^4 (simplified), which is 1 in 160,000. The probability of a specific 5-amino acid sequence arising by random chance on the same basis jumps to 1 in 3.2million!
So let’s hit the Protein BLAST button and away we go… and this is the screen you will get to, that I’m going to walk you through
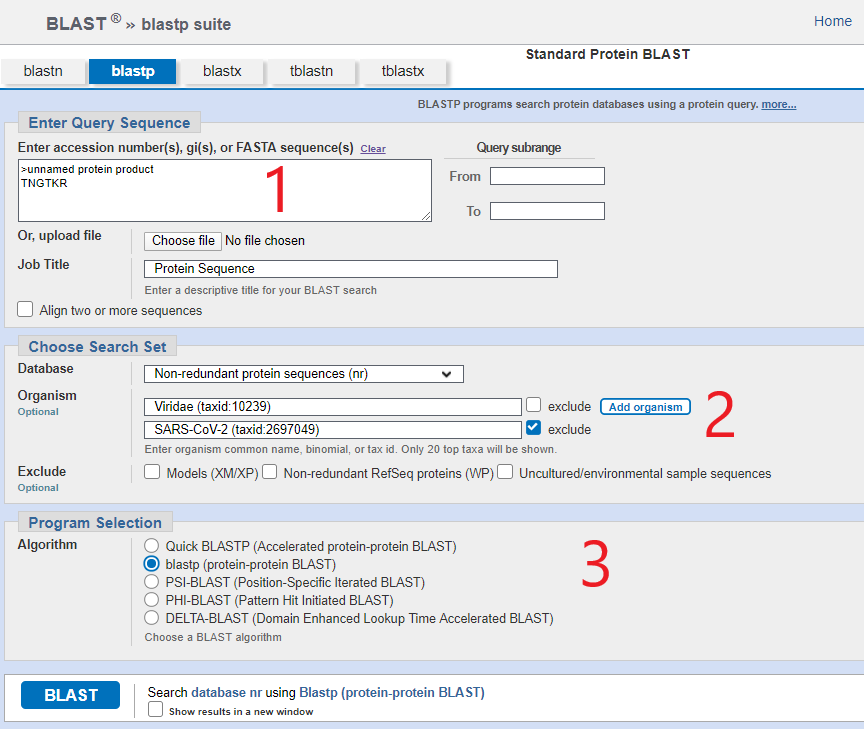
In [1] you need to enter your amino acid sequence of interest (BLAST adds “>unnamed protein product” automatically) . Fortunately you don’t need to look hard for this because we are going to concentrate on only 4 sequences within the SARS-CoV-2 viral genome/proteome and these are laid out for us in Prashant Pradhan’s wonderful paper “Uncanny similarity of unique inserts in the 2019-nCoV spike protein to HIV-1 gp120 and Gag” published 31st Jan 2020 a few days after the genome sequence was released.
The bit you need is in table 1 which I am reposting here and you will see that I have posted the 6-amino acid sequence TNGTKR in the box [1] marked in red onthe BLASTp screen.
For step [2] in the BLASTp entry screen you need to add some filters. The first filter is to restrict the search to “viridae (viruses)” (or you can just enter 10239 which is the taxonomy ID). The reason for this is that there are gazillions of species on the planet and BLASTp will search for all of them, but you really only want to know which virus this motif came from. You’re not really interested if the motif is found in a squid, although it is possible that a squid also got in on the action with the famous bat-and-pangolin tete-a-tete touted by the likes of Peter Daszak and Dominic Dwyer, making it a zoonotic menagerie-a-trois, but let’s stick to reality.
The second condition is to exclude all the references to SARS-CoV-2 that have now accumulated in the database, because those will all pop up (thousands of them) and we’re not interested.
Once you’ve entered those hit the BLAST button and what do you get? You will get a list of candidates that have close homology to that sequence. Because it is a very short sequence the homology (likeness) should be 100%. The top of the page will be a summary of what you have requested and the rest of the page is a list of the matching sequences. What you will see immediately is near the top of the list are two synthetic viruses which are a chimaera of SARS-Cov-2 and another virus, which have appeared in the last 2 years by labs making more viruses (because we don’t have enough). The next in the list is a whole bunch of references to HIV-1.
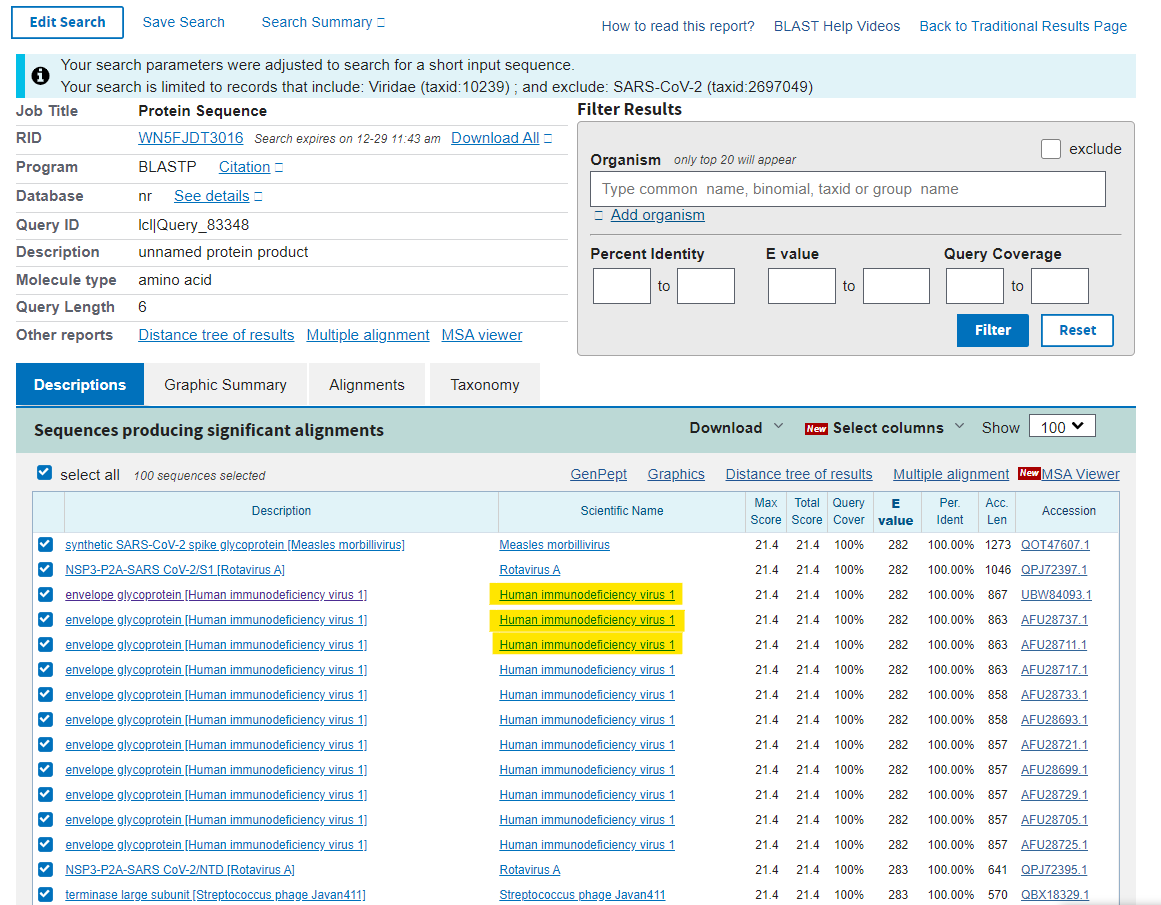
You can click on any of these and you will be taken to the alignment screen where the alignments between the subject (your TNGTKR) and query (all viruses) are shown, and you will see as you go down the page that the alignments only hold for HIV-1 until you start getting synthetic and hypothetical proteins, until the next real virus in the list which is HIV-2…
OK but one hit like this could be coincidental. In the list you will see the “E-value” which is an indicator of the probability of finding matches like this and should be as close to zero as possible. Here it’s 282 which really just reflects the likelihood of finding matches on a short sequence.
So, here’s the rub. Either these sequences are by chance going to match up with a whole bunch of other viruses (because the E-value is high and therefore we should expect a lot of matches) or they are really unusual sequences that have specifically, preferentially or uniquely match with HIV-1. How shall we address this? Well let’s go to the next sequence - HKNNKS, which is another short sequence. Just to tidy up the screen we can set a filter for short sequences to ensure that 100% of the sequence matches (top right). Now remember that if the match to HIV-1 was random, we shouldn’t really see a match on this list, because it should be bumped off by all the other hundreds of viruses that should be matching preferentially. Oops…
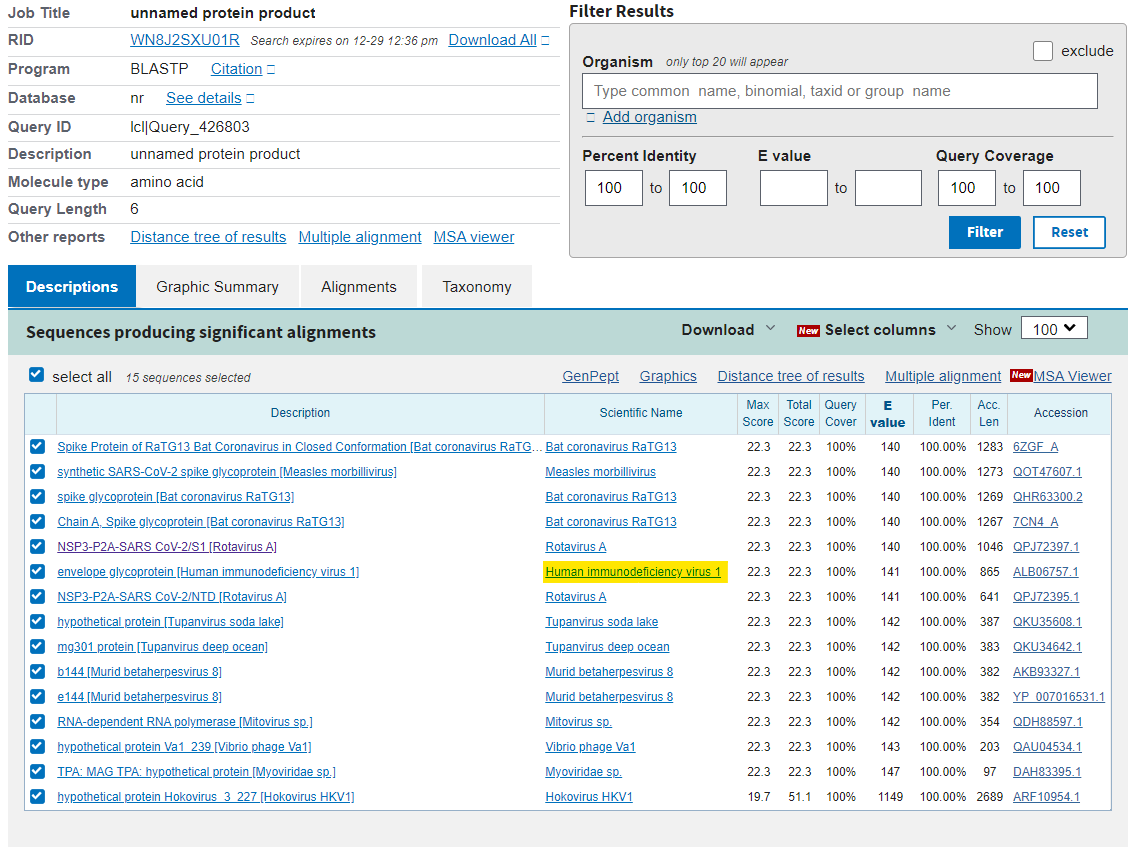
Note the other matches - Bat RaTG13 which didn’t appear in this database until after people started questioning the origin of the coronavirus, and is likely to be a synthetic sequence, and the same synthetic viruses that came after outbreak. So, HIV-1 is the ONLY match for both of these sequences.
Let’s go to sequence 3. This is a longer sequence. RSYLTPGDSSSG. I wonder what virus (or set of random viruses, because it’s a random sequence, remember) this will match to…. Oh look…
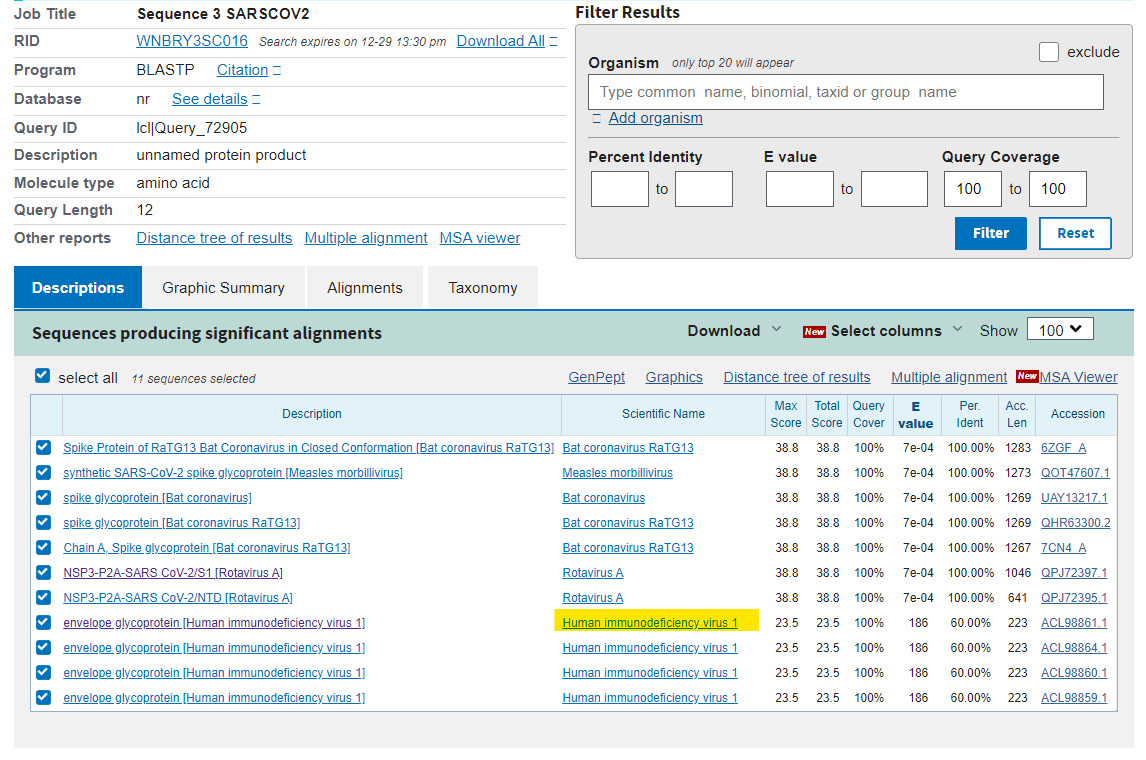
So in this search I have restricted the query coverage to 100% to get rid of the noise and the hypothetical proteins. All we are left with are the synthetic viruses from post-covid and RaTG13 (also post-covid). The only remaining virus in this list is, you guessed it, HIV-1. What are the odds that HIV-1 would pop up in all 3 searches?
And just to complete the quad-trick we need the last insert sequence identified in Pradhan’s paper which is QTNS——PRRA. This is a really interesting sequence which we will come to later, because is the furin cleavage site. It’s interesting because beta coronaviruses likes this don’t have a furin cleavage site, this is the only one. Surely this site couldn’t have come from HIV-1? Well, it’s not from the GP120 protein like the other three sequences, it’s completely different and on a different location of the virus which I’ll show you soon but for now let’s run the BLASTp.
This time it’s a bit messier because there have been a bunch of hypothetical and synthetic proteins added since SARS-CoV-2 was released (I should have written this piece last year). But HIV-1 makes its appearance on the list again and this time I’ll just show the alignments between the gag protein and the coronavirus - in this case there is a deletion from the HIV-1 protein.
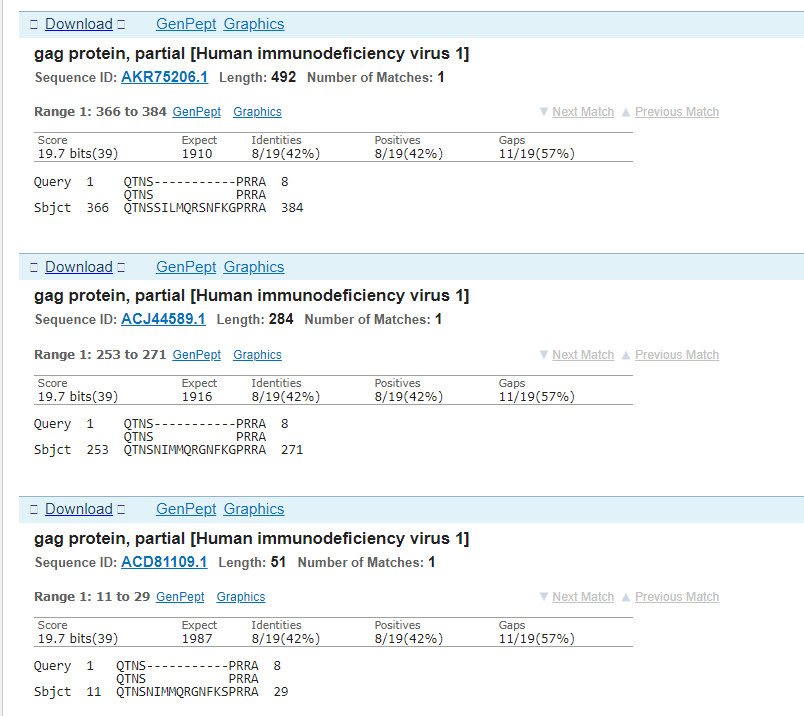
So, there we have 4 matches to HIV sequences with no other viruses* appearing in all 4 match lists (*barring synthetic ones created after the event). What are the odds of that - close to zero.
But look at this. These were not just random sequences from HIV. In his paper, Pradhan went further and recreated the structure of the virus with location of the four inserts. Lo and behold, these “random” inserts - all from HIV - are all at binding sites of the coronavirus. What are the odds?
Now, it’s possible that you aren’t convinced. Despite the fact that the only virus to appear in match lists for all four inserts, from the hundreds of thousands of viruses around, happens to be HIV-1. And HIV-1 should have no real chance of forming recombinant viruses with bat coronaviruses in nature, and no real chance of forming 4 different recombinations that just happen to be at binding sites for the virus. But if that doesn’t convince you there is one special feature of insert 4 we need to look at.
Now we are going back to nucleotides, the G-A-C-T’s that make up the sequence that code for the amino acids that we have so far been talking about. The original reference genome sequence for the coronavirus has a genbank ID of NC_045512.1 and can be seen here: https://www.ncbi.nlm.nih.gov/nuccore/NC_045512.1
You can play around with the genome sequence using BLASTn (for nucleotide) by going to that page and selecting “Run BLAST” from the right hand column which will take you to a similar BLAST page as had with the proteins above.
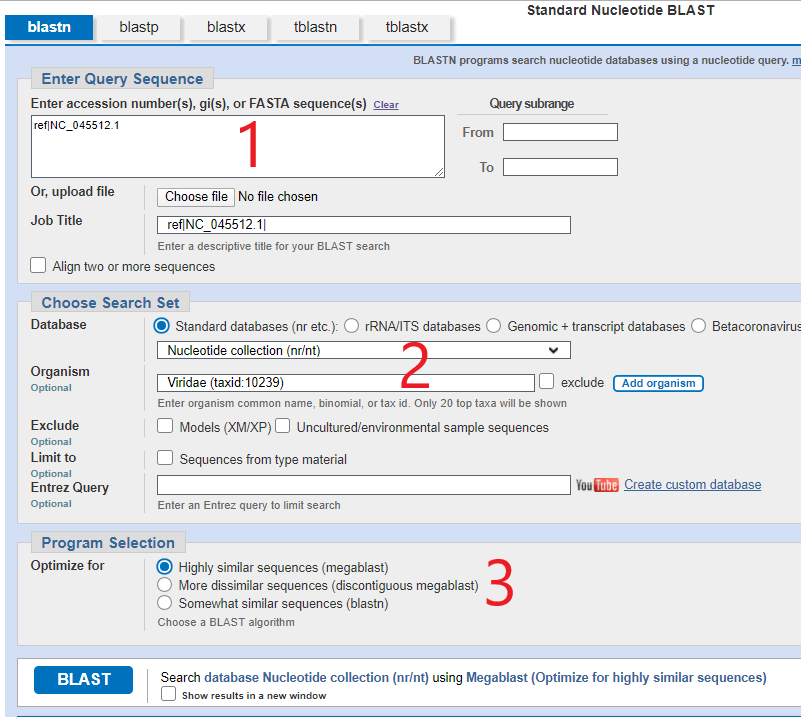
In a similar way you would enter NC_045512.1 (or the updated NC_045512.2) into the first box, choose the options shown in the part marked [2], select megablast in part 3 and click go, and you will get a list of SAR-CoV-2 genomes that match (obviously). I won’t show that screen because it’s not important here but this is the screen you get if you were to look at two closely matched sequences and you can click “CDS feature” to superimpose the amino acid sequence. You will end up with pages of something that looks like this:

In this particular section you can see it’s a sequence of the “spike protein” (surface glycoprotein) and the nucleotides are labelled 23548…23771 (of about 30,000 nucleotides or bases i.e. G-C-A-T). [NB: in actual fact this is RNA so should have a U in place of every T, but BLAST compensates for this automatically for simplicity]. The smaller number is the number of amino acid in the protein sequence so for each 3 nucleotides, the number goes up by 1 amino acid. The highlight is amino acid 677 (Q) to 686 (S), giving 677→686 = QTNSPRRARS.
Now, this is really interesting because not only have we seen that the QTNS section is derived from HIV but there is something very special about the adjacent PRRAR because that is a furin cleavage site and as we have seen, these don’t exist in this type of SARS-like virus. It’s an insertion to the viral genome, but nobody really knows how it got there (just like the HIV sequences). In order to see where it came from we need to look outside the amino acid sequence and back to the genome sequence.
The genome sequence that you can see for this amino acid sequence is: CAGACTAATTCTCCTCGGCGGGCACGTAGT which is 30 nucleotides coding for 10 amino acids. For this sequence to arise by chance would be an infinitesimally small number, so it has to have arisen somewhere (i.e. from another virus) or else some of it must be synthetic. So let’s BLAST(n) it, and this time we exclude “synthetic constructs” from our search (because we are looking for real viruses, not synthetic ones). What do we get?
So, now you are getting used to these displays we can see that the only viral sequences in here are synthetic, and if you were to click on each of these you would find their registration date after Feb 2020. In other words, no virus in existence has this genetic sequence. Well this is strange, because in order for a virus to acquire a large sequence like this it has to get it from another organism. It has no lab to manipulate gene sequences, neither do the bats (hence Jikky the lab mouse’s little joke)…

It’s easy enough to change a single nucleotide (a single point mutation or SNP) or even insert or delete nucleotides (less common) but to insert 20 or 30 nucleotides with a code that works? Nope, that has to come from another virus or else it’s been done in a lab.
So, where did this code come from? Well it turns out that BLAST can tell us - with some degree of certainty - where some of this code, particularly the bit that codes for the PRRAR section (the furin cleavage site which is so unique), came from.
The bit that we are interested in is in the clue from Jikky. CTCCTCGGCGGGCACGTAG. Let’s BLAST it
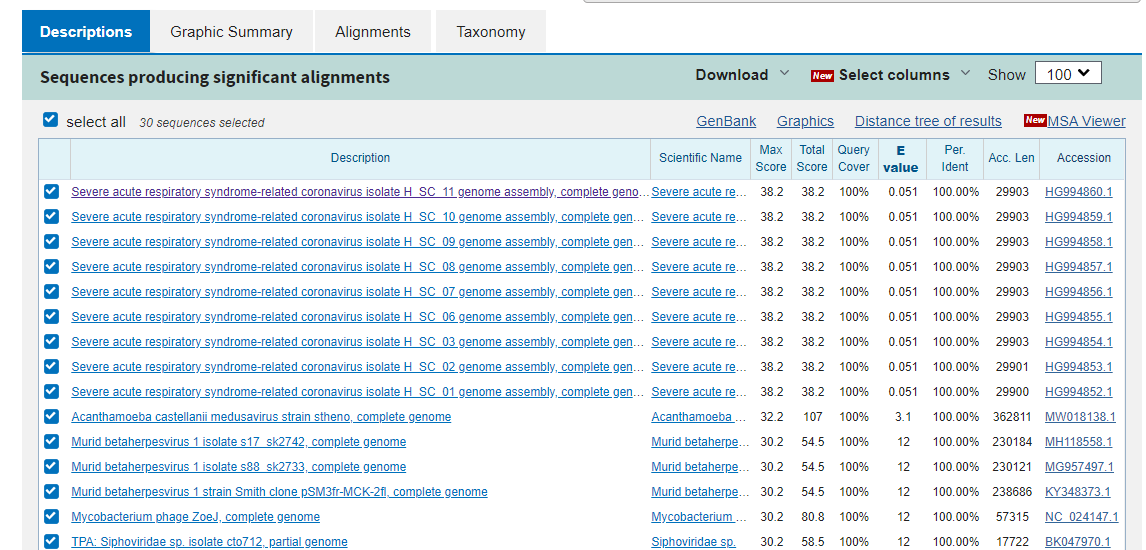
What you see is the same (misclassified SARS-CoV-2) sequences in the first 9 hits, and then none of the remaining hits have 19/19 matches. What this means is that there is no virus known to man that has this particular sequence in its genome prior to the discovery of SARS-Cov-2. So where on earth has it come from? For this you need to select a different database. Let’s go back to the BLASTn query screen and change the database option to “Patent sequences (pat)”. Remove all the exclusions and run the BLAST.

The results here need a little bit of sifting through because the top of the list include results from patents from this year. They are prefixed WO2021 and WO2020 so can be ignored. Just below those are the ones that we are interested in. I have just highlighted the top three but the whole list of many are patents owned by the same company, you just have to click on the accession number on the right.
So let’s do it and see which company, that we all know (now), that is a pharma company that has never produced a working drug yet has a market cap of over $80bn…
Yes, that’s right. Every single one of these patents that contains that 19nt sequence (for which the probability of occurring by random chance is less than 1 in a billion) is from Moderna. [Note the sequence is actually the reverse complement sequence but this is likely a direct result of the cell lines that it occurred in - MSH3_mutated cell lines designed for developing cancer vaccines, the Moderna patent was actually for a mutated MSH3 gene for this purpose]
In order for that sequence to have arisen in that virus, the virus which was manufactured with its HIV inserts, had to have had been infected into patented cell lines supplied by Moderna that had that unique sequence not seen in any other virus.
In theory nothing is impossible in science, medicine or genomics. A SARS virus emerging naturally with 3 HIV inserts at its binding sites and also containing a furin cleavage site that doesn’t exist in nature but does exist in a Moderna patent… that’s seriously crazy talk. It doesn’t exist. A flying pink elephant would be a million times more likely.
[UPDATE 21st Feb 2022… The paper we have been waiting to be released confirming the 19nt sequence in a Moderna patent as discussed above is now published. It’s worth a read if you have time]
[UPDATE 4th Dec 2022… I should have added links before to Charles Rixey’s extensive article on the documents underlying this “The myth of the blind watchmaker” as well as the follow-up article (part 2) “Absolute proof: The GP120 sequences” which are now added below, in addition to Igor Chudov’s excellent summary piece “Sars-Cov-2 was lab made under project DEFUSE”]


Taking a MSH3 mutated cell line and co transfecting it with a SARS like coronavirus cannot be called a "natural recombination event" in anyone's book.
This work should be the subject of a real congressional hearing. The data are damn near irrefutable. As a scientist, I am continually astounded that more scientists are not speaking up about this.